
In a remarkable discovery that underscores the rich biodiversity of the Great Barrier Reef, a team of marine biologists from the University of the Sunshine

Purchasing a redtail garra, an algae-eating fish, is as simple as a few mouse clicks. But obtaining details about its biology proves to be more

In the bustling urban expanse of Hangzhou City, situated in southeast China’s Zhejiang Province, an aquatic treasure has recently been unveiled. A new species of

When the genomes of 150,000 individuals were scanned, new genes linked to autism and certain other neuro-developmental disorders were identified. The next step will now
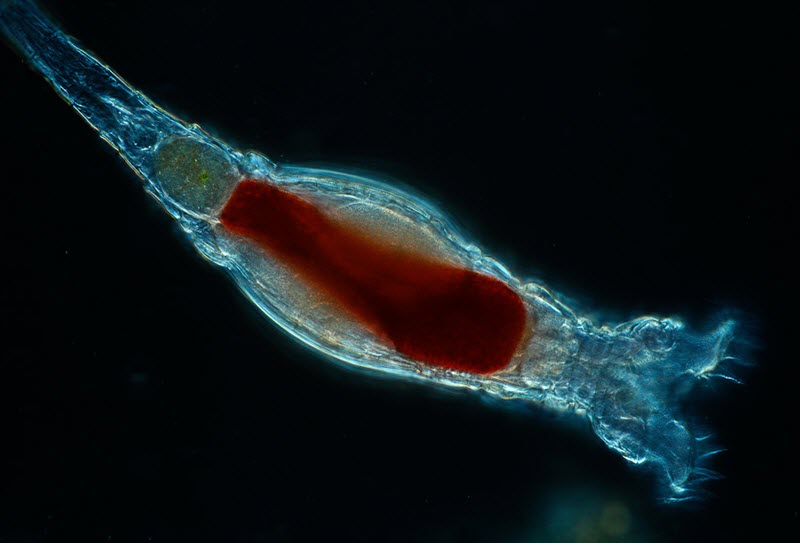
Scientists with the Marine Biological Laboratory have discovered a new form of genetic modification carried out by tiny animals called bdelloid rotifiers. This form of

The last recorded sighting of Maclear’s rat (Rattus macleari) was in 1903 and it is considered extinct, but scientists are now working to bring it back
A research team at Yale Cancer Center have discovered new consequences of specific gene mutations linked to the development of both myelodysplastic syndromes (MDS) and

A research team has been able to recover human DNA from the sticky substance that hair lice excrete to glue their eggs to the hairs

When there is a build-up of DNA-repair protein in the brain, the zebrafish (danio Rerio) will respond by sleeping, according to a new study on

According to a study published in September 2021, a team of researchers in China have found what may be DNA molecules from a 125-million-year-old dinosaur.